箱线图
箱线图是能同时反映数据统计量和整体分布,又很漂亮的展示图。在2014年的Nature Method上有2篇Correspondence论述了使用箱线图的好处和一个在线绘制箱线图的工具。就这样都可以发两篇Nature method,没天理,但也说明了箱线图的重要意义。
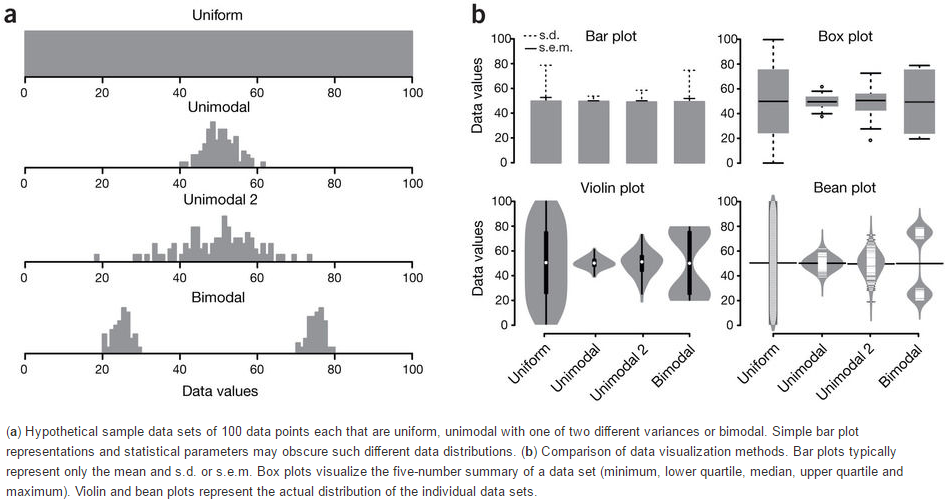
下面这张图展示了Bar plot、Box plot、Volin plot和Bean plot对数据分布的反应。从Bar plot上只能看到数据标准差或标准误不同;Box plot可以看到数据分布的集中性不同;Violin plot和Bean plot展示的是数据真正的分布,尤其是对Biomodal数据的展示。
Boxplot从下到上展示的是最小值,第一四分位数 (箱子的下边线)、中位数 (箱子中间的线)、第三四分位数 (箱子上边线)、最大值,具体解读参见 http://mp.weixin.qq.com/s/t3UTI_qAIi0cy1g6ZmHtwg。
- Nature Method文章 http://www.nature.com/nmeth/journal/v11/n2/full/nmeth.2811.html
一步步解析箱线图绘制
假设有这么一个基因表达矩阵,第一列为基因名字,后面几列为样品名字,想绘制下样品中基因表达的整体分布。
profile="Name;2cell_1;2cell_2;2cell_3;4cell_1;4cell_2;4cell_3;zygote_1;zygote_2;zygote_3
A;4;6;7;3.2;5.2;5.6;2;4;3
B;6;8;9;5.2;7.2;7.6;4;6;5
C;8;10;11;7.2;9.2;9.6;6;8;7
D;10;12;13;9.2;11.2;11.6;8;10;9
E;12;14;15;11.2;13.2;13.6;10;12;11
F;14;16;17;13.2;15.2;15.6;12;14;13
G;15;17;18;14.2;16.2;16.6;13;15;14
H;16;18;19;15.2;17.2;17.6;14;16;15
I;17;19;20;16.2;18.2;18.6;15;17;16
J;18;20;21;17.2;19.2;19.6;16;18;17
L;19;21;22;18.2;20.2;20.6;17;19;18
M;20;22;23;19.2;21.2;21.6;18;20;19
N;21;23;24;20.2;22.2;22.6;19;21;20
O;22;24;25;21.2;23.2;23.6;20;22;21"
读入数据并转换为ggplot2需要的长数据表格式 (经过前面几篇的练习,这应该都很熟了)
profile_text <- read.table(text=profile, header=T, row.names=1, quote="",sep=";", check.names=F)
# 在melt时保留位置信息
# melt格式是ggplot2画图最喜欢的格式
# 好好体会下这个格式,虽然多占用了不少空间,但是确实很方便
library(ggplot2)
library(reshape2)
data_m <- melt(profile_text)
head(data_m)
variable value
1 2cell_1 4
2 2cell_1 6
3 2cell_1 8
4 2cell_1 10
5 2cell_1 12
6 2cell_1 14
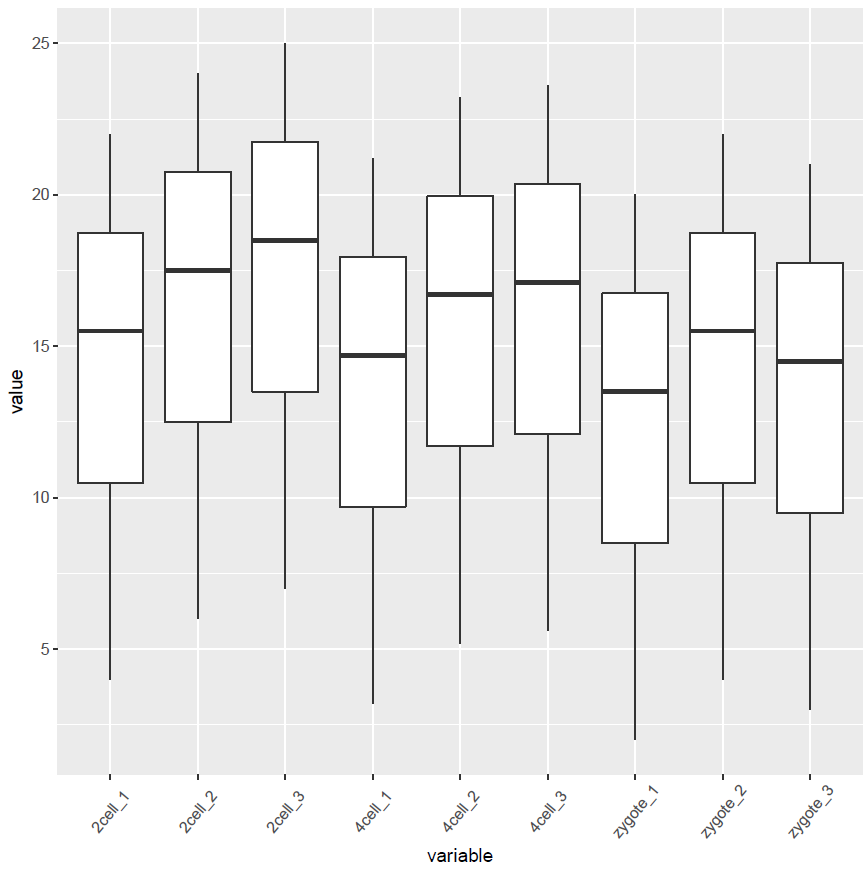
像往常一样,就可以直接画图了。
# variable和value为矩阵melt后的两列的名字,内部变量, variable代表了点线的属性,value代表对应的值。
p <- ggplot(data_m, aes(x=variable, y=value),color=variable) +
geom_boxplot() +
theme(axis.text.x=element_text(angle=50,hjust=0.5, vjust=0.5)) +
theme(legend.position="none")
p
# 图会存储在当前目录的Rplots.pdf文件中,如果用Rstudio,可以不运行dev.off()
dev.off()
箱线图出来了,看上去还可以,再加点色彩。
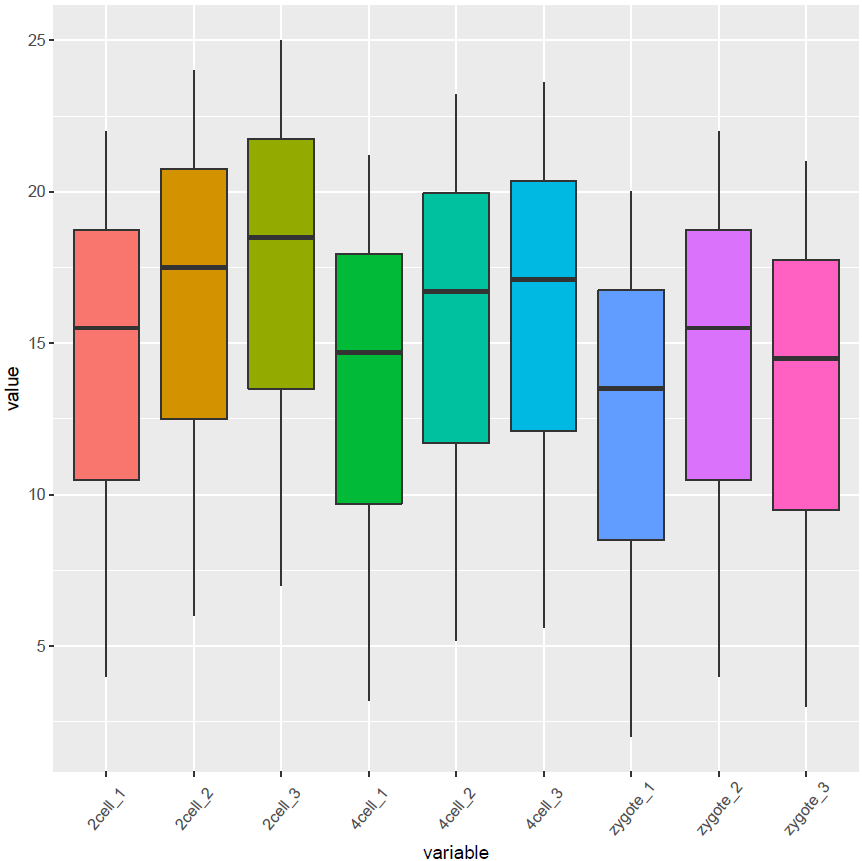
# variable和value为矩阵melt后的两列的名字,内部变量, variable代表了点线的属性,value代表对应的值。
p <- ggplot(data_m, aes(x=variable, y=value),color=variable) +
geom_boxplot(aes(fill=factor(variable))) +
theme(axis.text.x=element_text(angle=50,hjust=0.5, vjust=0.5)) +
theme(legend.position="none")
p
# 图会存储在当前目录的Rplots.pdf文件中,如果用Rstudio,可以不运行dev.off()
dev.off()
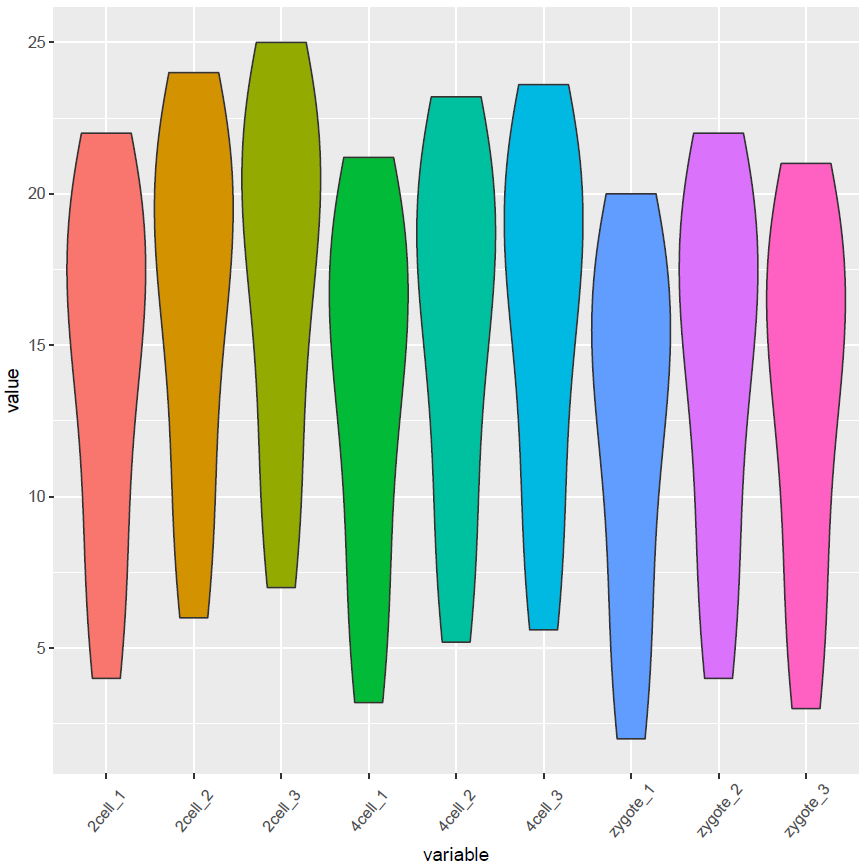
再看看Violin plot
# variable和value为矩阵melt后的两列的名字,内部变量, variable代表了点线的属性,value代表对应的值。
p <- ggplot(data_m, aes(x=variable, y=value),color=variable) +
geom_violin(aes(fill=factor(variable))) +
theme(axis.text.x=element_text(angle=50,hjust=0.5, vjust=0.5)) +
theme(legend.position="none")
p
# 图会存储在当前目录的Rplots.pdf文件中,如果用Rstudio,可以不运行dev.off()
dev.off()
还有Jitter plot (这里使用的是ggbeeswarm包)
library(ggbeeswarm)
# 为了更好的效果,只保留其中一个样品的数据
# grepl类似于Linux的grep命令,获取特定模式的字符串
data_m2 <- data_m[grepl("_3", data_m$variable),]
# variable和value为矩阵melt后的两列的名字,内部变量, variable代表了点线的属性,value代表对应的值。
p <- ggplot(data_m2, aes(x=variable, y=value),color=variable) +
geom_quasirandom(aes(colour=factor(variable))) +
theme_bw() + theme(panel.grid.major = element_blank(),
panel.grid.minor = element_blank(), legend.key=element_blank()) +
theme(legend.position="none")
# 也可以用geom_jitter(aes(colour=factor(variable)))代替geom_quasirandom(aes(colour=factor(variable)))
# 但个人认为geom_quasirandom给出的结果更有特色
ggsave(p, filename="jitterplot.pdf", width=14, height=8, units=c("cm"))
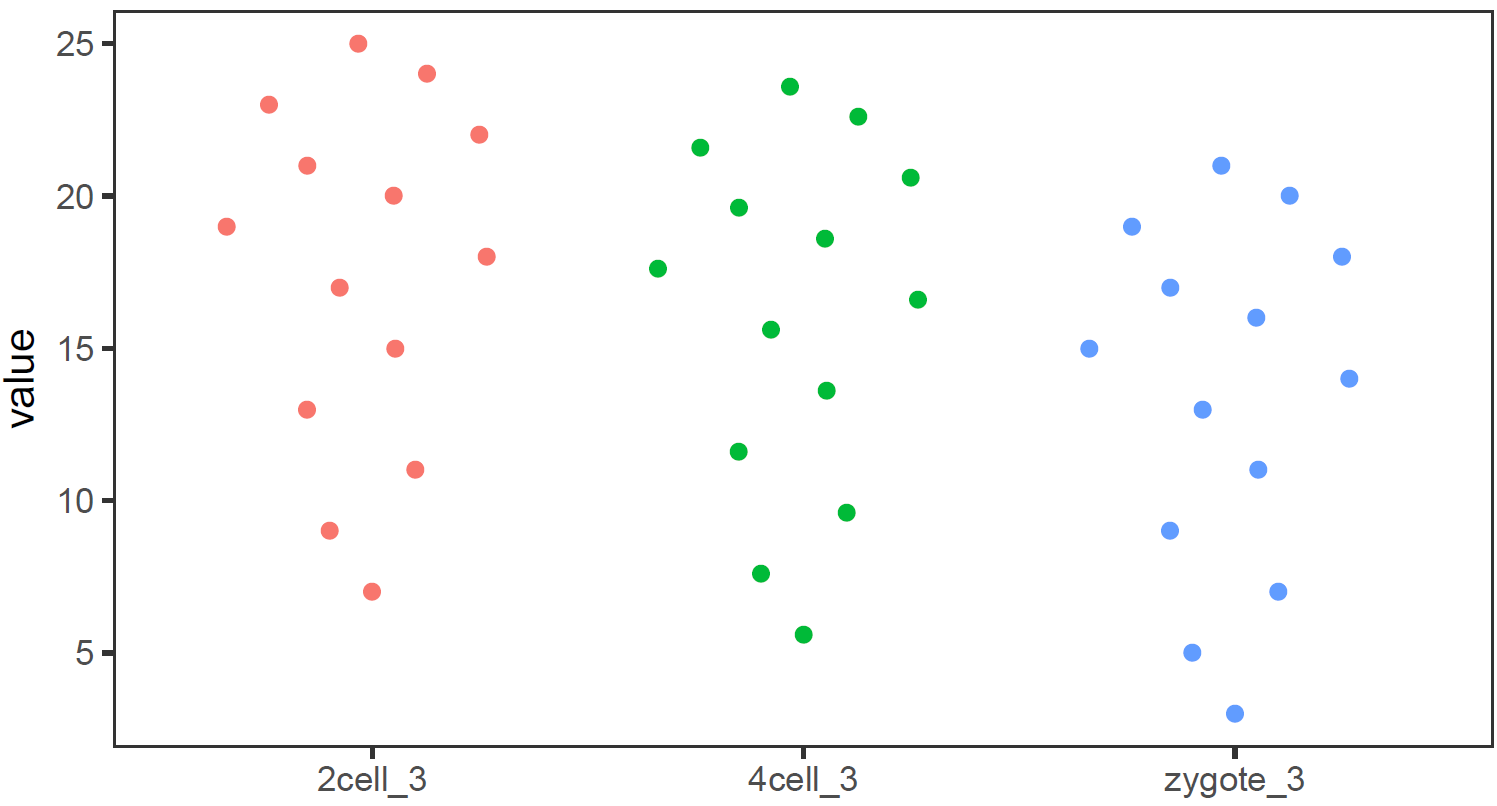
绘制单个基因 (A)的箱线图
为了更好的展示效果,下面的矩阵增加了样品数量和样品的分组信息。
profile="Name;2cell_1;2cell_2;2cell_3;2cell_4;2cell_5;2cell_6;4cell_1;4cell_2;4cell_3;4cell_4;4cell_5;4cell_6;zygote_1;zygote_2;zygote_3;zygote_4;zygote_5;zygote_6
A;4;6;7;5;8;6;3.2;5.2;5.6;3.6;7.6;4.8;2;4;3;2;4;2.5
B;6;8;9;7;10;8;5.2;7.2;7.6;5.6;9.6;6.8;4;6;5;4;6;4.5"
profile_text <- read.table(text=profile, header=T, row.names=1, quote="",sep=";", check.names=F)
data_m = data.frame(t(profile_text['A',]))
data_m$sample = rownames(data_m)
# 只挑选显示部分
# grepl前面已经讲过用于匹配
data_m[grepl('_[123]', data_m$sample),]
A sample
2cell_1 4.0 2cell_1
2cell_2 6.0 2cell_2
2cell_3 7.0 2cell_3
4cell_1 3.2 4cell_1
4cell_2 5.2 4cell_2
4cell_3 5.6 4cell_3
zygote_1 2.0 zygote_1
zygote_2 4.0 zygote_2
zygote_3 3.0 zygote_3
获得样品分组信息 (这个例子比较特殊,样品的分组信息就是样品名字下划线前面的部分)
# 可以利用strsplit分割,取出其前面的字符串
# R中复杂的输出结果多数以列表的形式体现,在之前的矩阵操作教程中
# 提到过用str函数来查看复杂结果的结构,并从中获取信息
group = unlist(lapply(strsplit(data_m$sample,"_"), function(x) x[1]))
data_m$group = group
data_m[grepl('_[123]', data_m$sample),]
A sample group
2cell_1 4.0 2cell_1 2cell
2cell_2 6.0 2cell_2 2cell
2cell_3 7.0 2cell_3 2cell
4cell_1 3.2 4cell_1 4cell
4cell_2 5.2 4cell_2 4cell
4cell_3 5.6 4cell_3 4cell
zygote_1 2.0 zygote_1 zygote
zygote_2 4.0 zygote_2 zygote
zygote_3 3.0 zygote_3 zygote
如果没有这个规律,也可以提到类似于下面的文件,指定样品所属的组的信息。
sampleGroup_text="Sample;Group
zygote_1;zygote
zygote_2;zygote
zygote_3;zygote
zygote_4;zygote
zygote_5;zygote
zygote_6;zygote
2cell_1;2cell
2cell_2;2cell
2cell_3;2cell
2cell_4;2cell
2cell_5;2cell
2cell_6;2cell
4cell_1;4cell
4cell_2;4cell
4cell_3;4cell
4cell_4;4cell
4cell_5;4cell
4cell_6;4cell"
#sampleGroup = read.table(text=sampleGroup_text,sep="\t",header=1,check.names=F,row.names=1)
#data_m <- merge(data_m, sampleGroup, by="row.names")
# 会获得相同的结果,脚本注释掉了以免重复执行引起问题。
矩阵准备好了,开始画图了 (小提琴图做例子,其它类似)
# 调整下样品出现的顺序
data_m$group <- factor(data_m$group, levels=c("zygote","2cell","4cell"))
# group和A为矩阵中两列的名字,group代表了值的属性,A代表基因A对应的表达值。
# 注意看修改了的地方
p <- ggplot(data_m, aes(x=group, y=A),color=group) +
geom_violin(aes(fill=factor(group))) +
theme(axis.text.x=element_text(angle=50,hjust=0.5, vjust=0.5)) +
theme(legend.position="none")
p
# 图会存储在当前目录的Rplots.pdf文件中,如果用Rstudio,可以不运行dev.off()
dev.off()
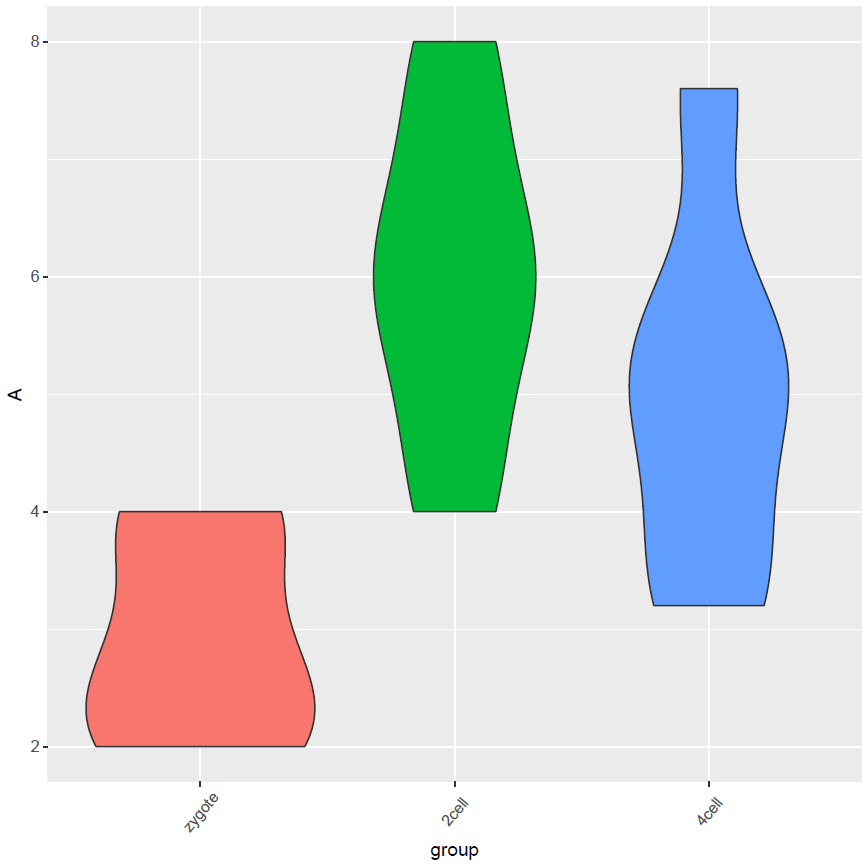
长矩阵绘制箱线图
常规矩阵绘制箱线图要求必须是个方正的矩阵输入,而有时想比较的几个组里面检测的值数目不同。比如有三个组,GrpA组检测了6个病人,GrpB组检测了10个病人,GrpC组是12个正常人的检测数据。这时就很难形成一个行位检测值,列为样品的矩阵,长表格模式就适合与这种情况。
long_table <- "Grp;Value
GrpA;10
GrpA;11
GrpA;12
GrpB;5
GrpB;4
GrpB;3
GrpB;2
GrpC;2
GrpC;3"
long_table <- read.table(text=long_table,sep="\t",header=1,check.names=F)
p <- ggplot(data_m, aes(x=Grp, y=Value),color=Grp) +
geom_violin(aes(fill=factor(Grp))) +
theme(axis.text.x=element_text(angle=50,hjust=0.5, vjust=0.5)) +
theme(legend.position="none")
p
长表格形式自身就是常规矩阵melt后的格式,这种用来绘制箱线图就很简单了,就不举例子了。
箱线图 - 一步绘制
绘图时通常会碰到两个头疼的问题:
-
有时需要绘制很多的图,唯一的不同就是输入文件,其它都不需要修改。如果用R脚本,需要反复替换文件名,繁琐又容易出错。 (R也有命令行参数,不熟,有经验的可以尝试下)
-
每次绘图都需要不断的调整参数,时间久了不用,就忘记参数怎么设置了;或者调整次数过多,有了很多版本,最后不知道用哪个了。
为了简化绘图、维持脚本的一致,我用bash对绘图命令做了一个封装,通过配置修改命令行参数,生成相应的绘图脚本,然后再绘制。
首先把测试数据存储到文件中方便调用。数据矩阵存储在boxplot.normal.data、sampleGroup和boxplot.melt.data文件中 (TAB键分割,内容在文档最后。如果你手上有自己的数据,也可以拿来用)。
使用正常矩阵默认参数绘制箱线图
# -f: 指定输入的矩阵文件,第一列为行名字,第一行为header
列数不限,列名字不限;行数不限,行名字默认为文本
sp_boxplot.sh -f boxplot.normal.data
箱线图出来了,但有点小乱。
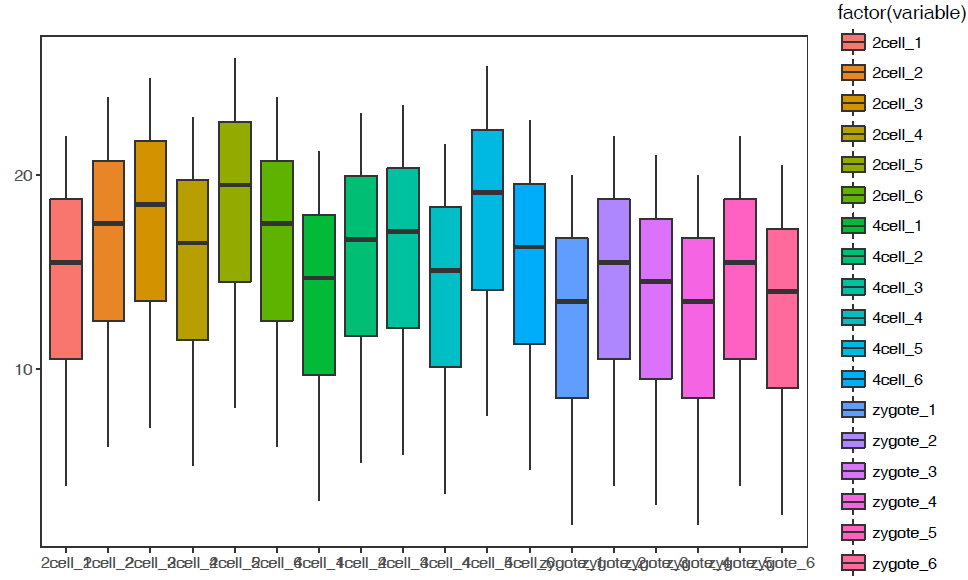
# -f: 指定输入的矩阵文件,第一列为行名字,第一行为header
列数不限,列名字不限;行数不限,行名字默认为文本
# -P: none, 去掉legend (uppercase P)
# -b: X-axis旋转45度
# -V: TRUE 绘制小提琴图
sp_boxplot.sh -f boxplot.normal.data -P none -b 45 -V TRUE
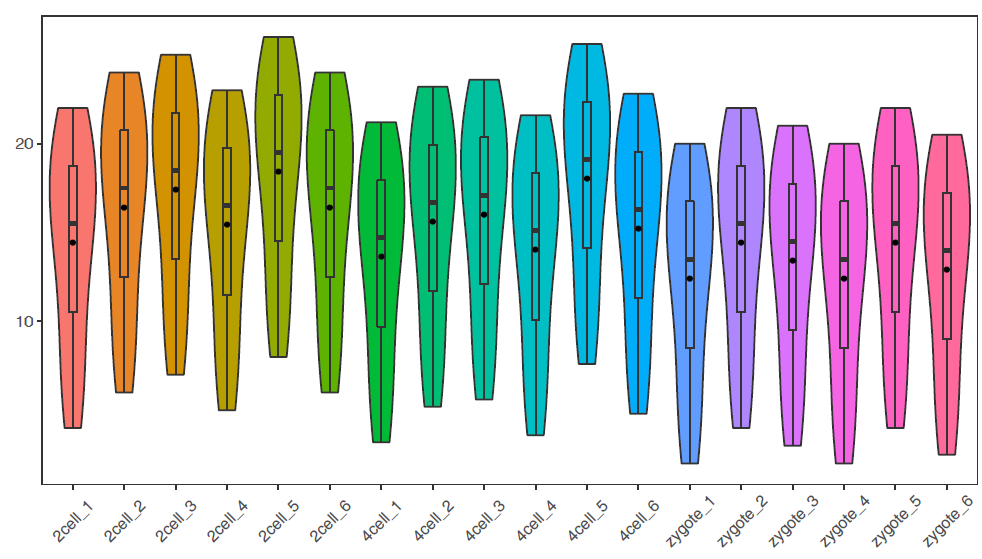
绘制单个基因的小提琴图加抖动图
# -q: 指定某一行的名字,此处为基因名,绘制基因A的表达图谱
# -Q: 指定样本分组,绘制基因A在不同样品组的表达趋势
# -F Group: sampleGroup中第二列的名字,指代分组信息,根据需要修改
# -J TRUE: 绘制抖动图 jitter plot
# -L: 设置X轴样品组顺序
# -c TRUE -C "'red', 'pink', 'blue'": 指定每个箱线图的颜色
sp_boxplot.sh -f boxplot.normal.data -q A -Q sampleGroup -F Group -V TRUE -J TRUE -L "'zygote','2cell','4cell'" -c TRUE -C "'red', 'pink', 'blue'" -P none
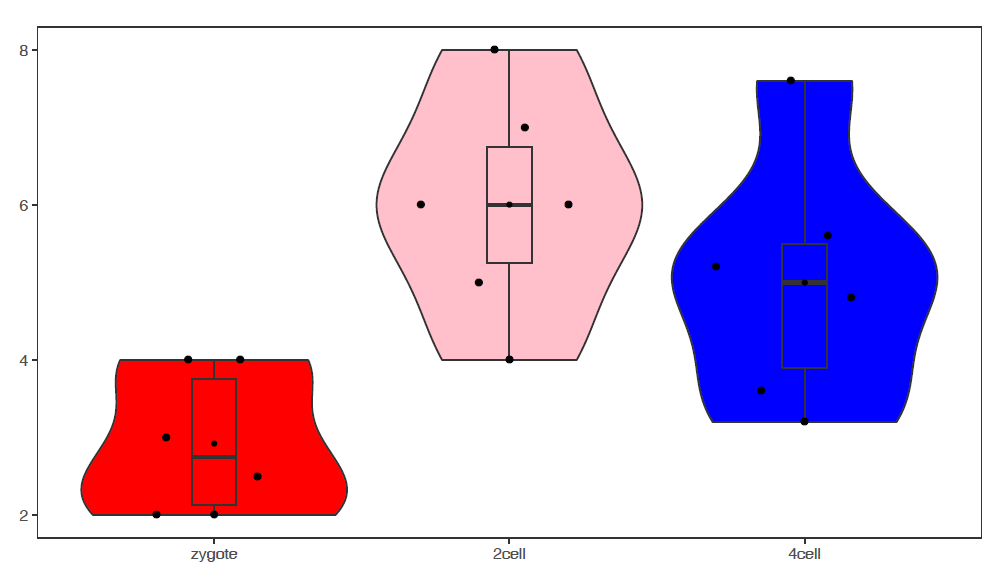
使用melted矩阵默认参数绘箱线图
# -f: 指定输入文件
# -m TRUE: 指定输入的矩阵为melted format
# -d Expr:指定表达值所在的列
# -F Rep: 指定子类所在列,也就是legend
# -a Group:指定X轴分组信息
# -j TRUE: jitter plot
sp_boxplot.sh -f boxplot.melt.data -m TRUE -d Expr -F Rep -a Group -j TRUE
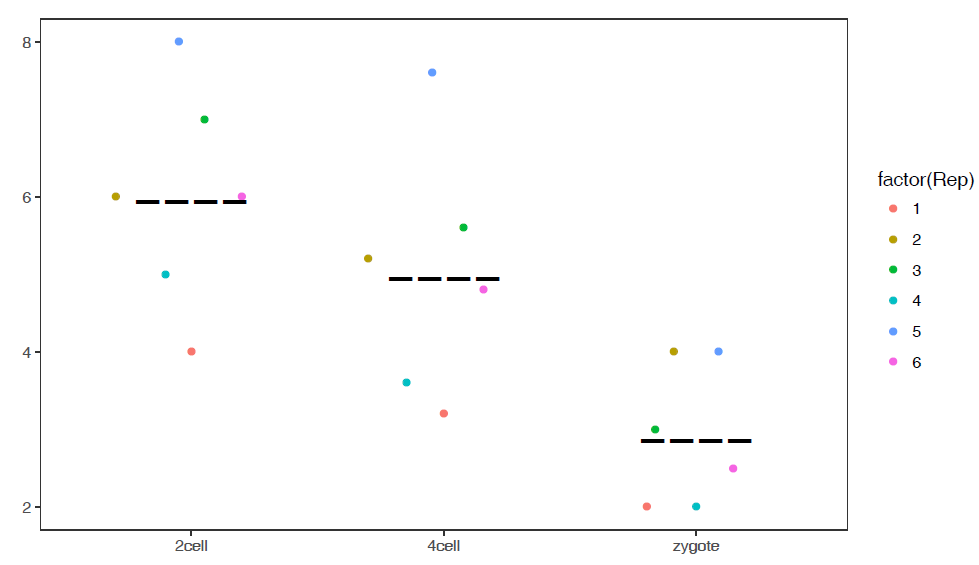
# 如果没有子类,则-a和-F指定为同一值
# -R TRUE: 旋转boxplot
sp_boxplot.sh -f boxplot.melt.data -m TRUE -d Expr -a Group -F Group -J TRUE -R TRUE
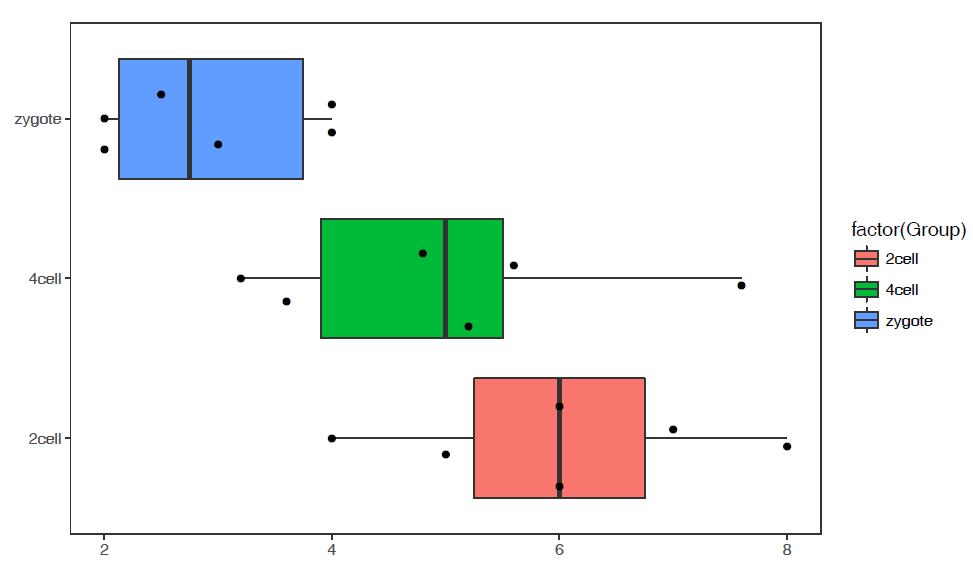
参数中最需要注意的是引号的使用:
- 外层引号与内层引号不能相同
- 凡参数值中包括了
空格,括号,逗号等都用引号括起来作为一个整体。
为了推广,也为了激起大家的热情,如果想要sp_boxplot.sh脚本的,还需要劳烦大家动动手,转发此文章到朋友圈,并留言索取。
也希望大家能一起开发,完善功能。
#boxplot.normal.data
Name 2cell_1 2cell_2 2cell_3 2cell_4 2cell_5 2cell_6 4cell_1 4cell_2 4cell_3 4cell_4 4cell_5 4cell_6 zygote_1 zygote_2 zygote_3 zygote_4 zygote_5 zygote_6
A 4 6 7 5 8 6 3.2 5.2 5.6 3.6 7.6 4.8 2 4 3 2 4 2.5
B 6 8 9 7 10 8 5.2 7.2 7.6 5.6 9.6 6.8 4 6 5 4 6 4.5
C 8 10 11 9 12 10 7.2 9.2 9.6 7.6 11.6 8.8 6 8 7 6 8 6.5
D 10 12 13 11 14 12 9.2 11.2 11.6 9.6 13.6 10.8 8 10 9 8 10 8.5
E 12 14 15 13 16 14 11.2 13.2 13.6 11.6 15.6 12.8 10 12 11 10 12 10.5
F 14 16 17 15 18 16 13.2 15.2 15.6 13.6 17.6 14.8 12 14 13 12 14 12.5
G 15 17 18 16 19 17 14.2 16.2 16.6 14.6 18.6 15.8 13 15 14 13 15 13.5
H 16 18 19 17 20 18 15.2 17.2 17.6 15.6 19.6 16.8 14 16 15 14 16 14.5
I 17 19 20 18 21 19 16.2 18.2 18.6 16.6 20.6 17.8 15 17 16 15 17 15.5
J 18 20 21 19 22 20 17.2 19.2 19.6 17.6 21.6 18.8 16 18 17 16 18 16.5
L 19 21 22 20 23 21 18.2 20.2 20.6 18.6 22.6 19.8 17 19 18 17 19 17.5
M 20 22 23 21 24 22 19.2 21.2 21.6 19.6 23.6 20.8 18 20 19 18 20 18.5
N 21 23 24 22 25 23 20.2 22.2 22.6 20.6 24.6 21.8 19 21 20 19 21 19.5
O 22 24 25 23 26 24 21.2 23.2 23.6 21.6 25.6 22.8 20 22 21 20 22 20.5
#boxplot.melt.data
Gene Sample Group Expr Rep
A zygote_1 zygote 2 1
A zygote_2 zygote 4 2
A zygote_3 zygote 3 3
A zygote_4 zygote 2 4
A zygote_5 zygote 4 5
A zygote_6 zygote 2.5 6
A 2cell_1 2cell 4 1
A 2cell_2 2cell 6 2
A 2cell_3 2cell 7 3
A 2cell_4 2cell 5 4
A 2cell_5 2cell 8 5
A 2cell_6 2cell 6 6
A 4cell_1 4cell 3.2 1
A 4cell_2 4cell 5.2 2
A 4cell_3 4cell 5.6 3
A 4cell_4 4cell 3.6 4
A 4cell_5 4cell 7.6 5
A 4cell_6 4cell 4.8 6
#sampleGroup
Sample Group
zygote_1 zygote
zygote_2 zygote
zygote_3 zygote
zygote_4 zygote
zygote_5 zygote
zygote_6 zygote
2cell_1 2cell
2cell_2 2cell
2cell_3 2cell
2cell_4 2cell
2cell_5 2cell
2cell_6 2cell
4cell_1 4cell
4cell_2 4cell
4cell_3 4cell
4cell_4 4cell
4cell_5 4cell
4cell_6 4cell
Reference
- http://blog.genesino.com//2017/06/R-Rstudio