柱状图绘制
柱状图也是较为常见的一种数据展示方式,可以展示基因的表达量,也可以展示GO富集分析结果,基因注释数据等。39个转录组分析工具,120种组合评估(转录组分析工具哪家强-导读版)中提到了较多堆积柱状图的使用。下面就详细介绍下怎么绘制。
常规矩阵柱状图绘制
有如下4个基因在5组样品中的表达值
data_ori <- "Grp_1;Grp_2;Grp_3;Grp_4;Grp_5
a;2.6;2.9;2.1;2.0;2.2
b;20.8;9.8;7.0;3.7;19.2
c;10.0;11.0;9.2;12.4;9.6
d;9;3.3;10.3;11.1;10"
data <- read.table(text=data_ori, header=T, row.names=1, sep=";", quote="")
data
Grp_1 Grp_2 Grp_3 Grp_4 Grp_5
a 2.6 2.9 2.1 2.0 2.2
b 20.8 9.8 7.0 3.7 19.2
c 10.0 11.0 9.2 12.4 9.6
d 9.0 3.3 10.3 11.1 10.0
整理数据格式,保留基因名字信息
library(ggplot2)
library(reshape2)
library(dplyr)
data_rownames <- rownames(data)
data_colnames <- colnames(data)
data$gene <- data_rownames
data_m <- melt(data, id.vars=c("gene"))
data_m
gene variable value
1 a Grp_1 2.6
2 b Grp_1 20.8
3 c Grp_1 10.0
4 d Grp_1 9.0
5 a Grp_2 2.9
6 b Grp_2 9.8
7 c Grp_2 11.0
8 d Grp_2 3.3
首先看下每个基因在不同组的表达情况
# 给定数据,和x轴、y轴所在列名字
# 直接使用geom_bar就可以绘制柱状图
# position: dodge: 柱子并排放置
p <- ggplot(data_m, aes(x=gene, y=value))
p + geom_bar(stat="identity", position="dodge", aes(fill=variable))
# 如果没有图形界面,运行下面的语句把图存在工作目录下的Rplots.pdf文件中
#dev.off()
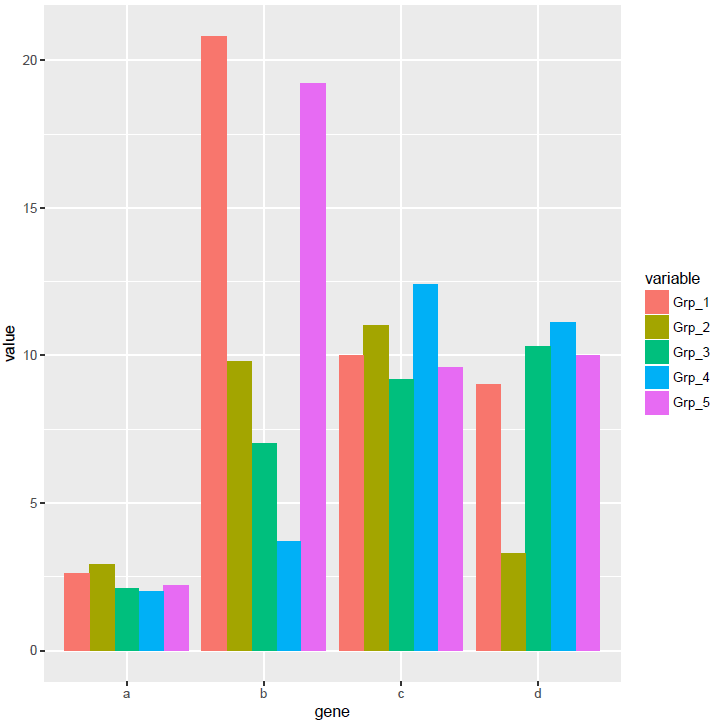
柱子有点多,也可以利用mean±SD的形式展现
首先计算平均值和标准差,使用group_by按gene分组,对每组做summarize
# 获取平均值和标准差
data_m_sd_mean <- data_m %>% group_by(gene) %>% dplyr::summarise(sd=sd(value), value=mean(value))
data_m_sd_mean <- as.data.frame(data_m_sd_mean)
data_m_sd_mean
gene sd value
1 a 0.3781534 2.36
2 b 7.5491721 12.10
3 c 1.2837445 10.44
4 d 3.1325708 8.74
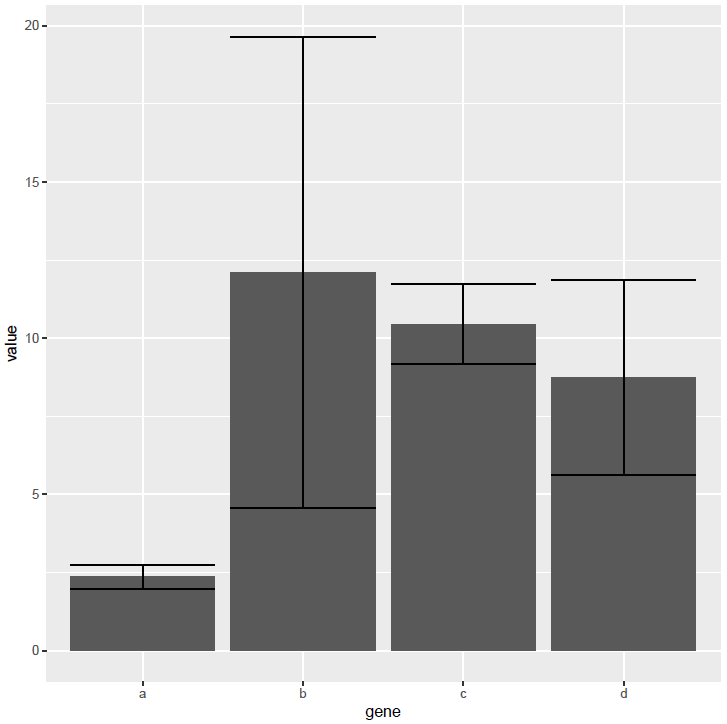
使用geom_errorbar添加误差线
p <- ggplot(data_m_sd_mean, aes(x=gene, y=value)) +
geom_bar(stat="identity") +
geom_errorbar(aes(ymin=value-sd, ymax=value+sd))
p
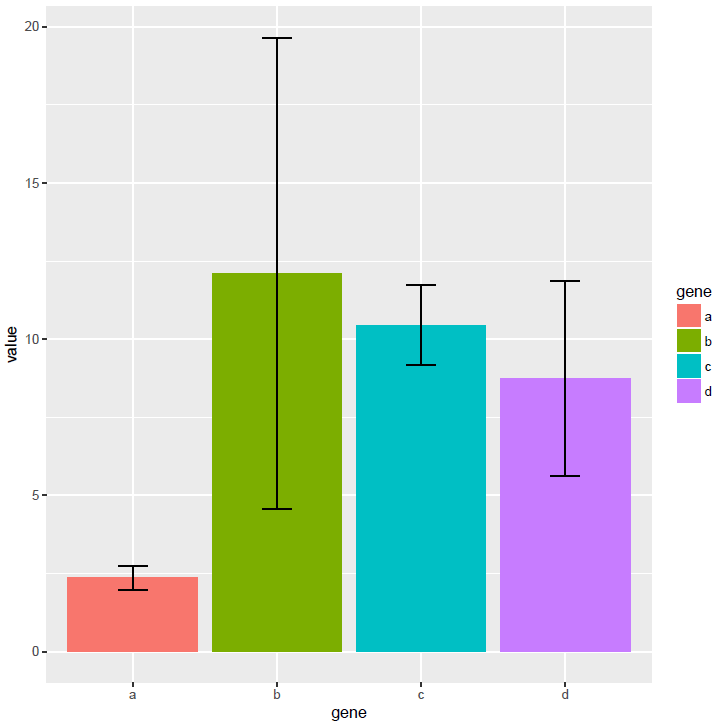
设置误差线的宽度和位置
p <- ggplot(data_m_sd_mean, aes(x=gene, y=value)) +
geom_bar(stat="identity", aes(fill=gene)) +
geom_errorbar(aes(ymin=value-sd, ymax=value+sd), width=0.2, position=position_dodge(width=0.75))
p
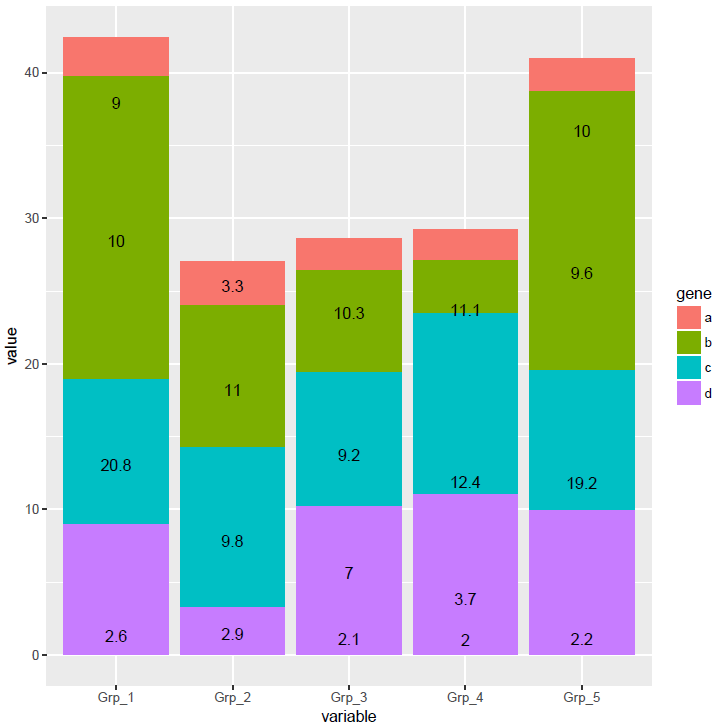
每个基因的原始表达值堆积柱状图 (只需要修改positon=stack)
# position="fill" 展示的是堆积柱状图各部分的相对比例
# position="stack" 展示的是堆积柱状图的原始值
p <- ggplot(data_m, aes(x=variable, y=value)) +
geom_bar(stat="identity", position="stack", aes(fill=gene)) +
geom_text(aes(label=value), position=position_stack(vjust=0.5))
p
堆积柱状图显示没问题,但文本标记错位了
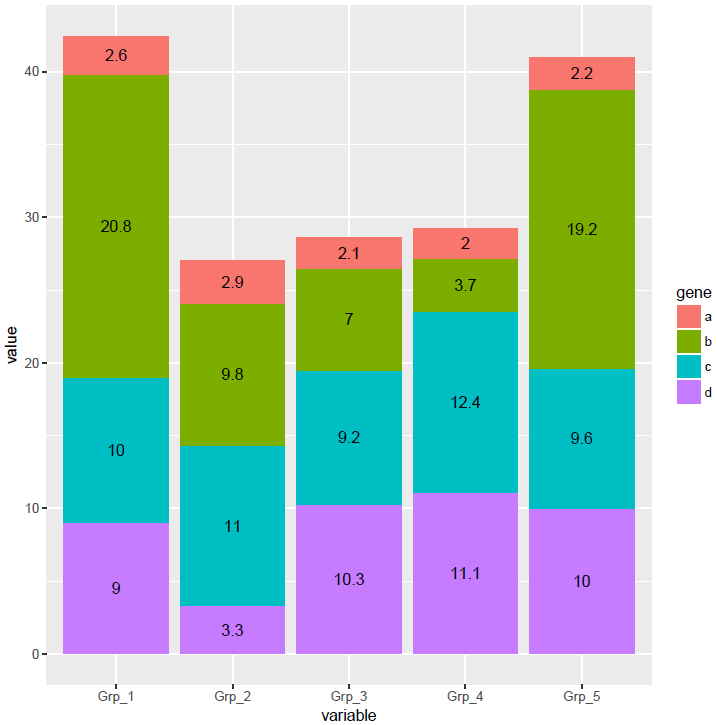
指定下分组信息,位置计算就正确了
# position="fill" 展示的是堆积柱状图各部分的相对比例
# position="stack" 展示的是堆积柱状图的原始值
p <- ggplot(data_m, aes(x=variable, y=value, group=gene)) +
geom_bar(stat="identity", position="stack", aes(fill=gene)) +
geom_text(aes(label=value), position=position_stack(vjust=0.5))
p
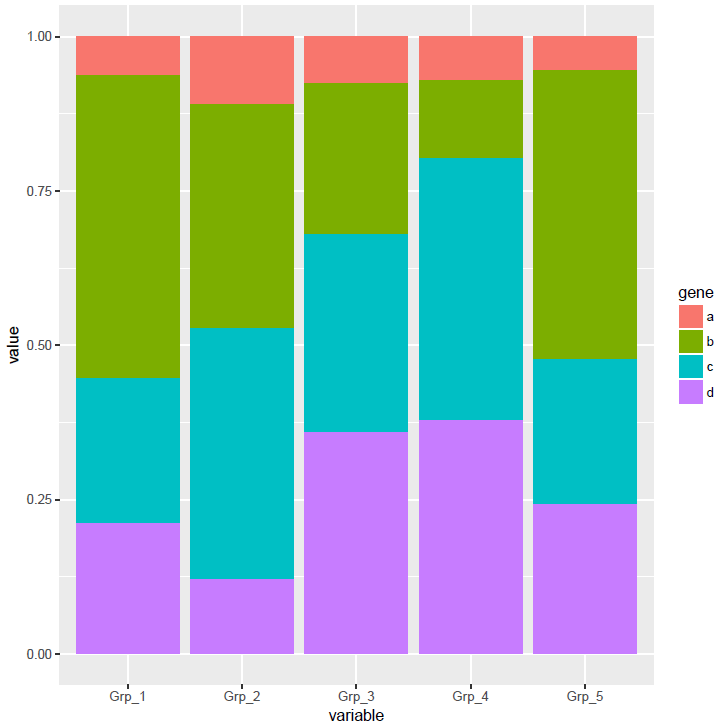
比较每组各个基因的相对表达 (position=fill)
# position="fill" 展示的是堆积柱状图各部分的相对比例
# position="stack" 展示的是堆积柱状图的原始值,可以自己体现下看卡差别
p <- ggplot(data_m, aes(x=variable, y=value)) +
geom_bar(stat="identity", position="fill", aes(fill=gene))
p
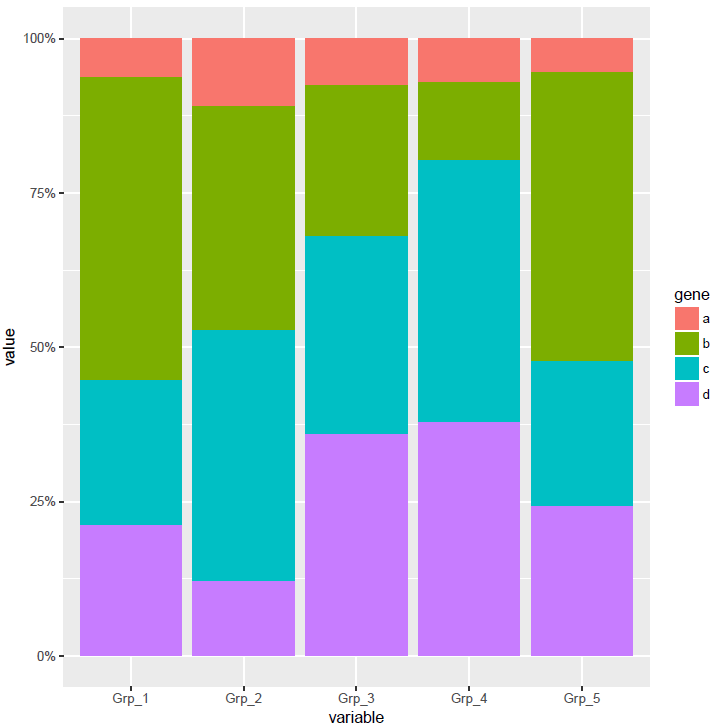
纵轴的显示改为百分比
p <- ggplot(data_m, aes(x=variable, y=value)) +
geom_bar(stat="identity", position="fill", aes(fill=gene)) +
scale_y_continuous(labels = scales::percent)
p
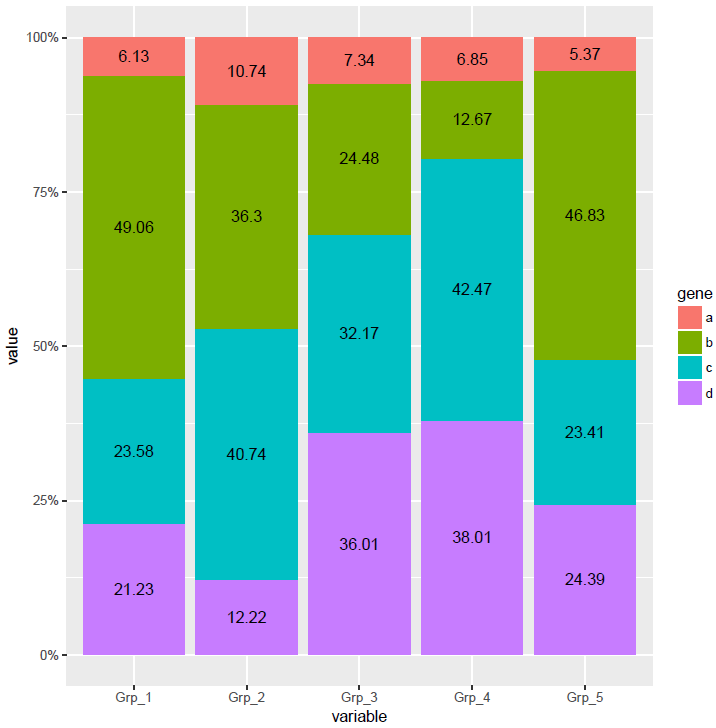
在柱子中标记百分比值
首先计算百分比,同样是group_by (按照给定的变量分组,然后按组操作)和mutate两个函数(在当前数据表增加新变量)
# group_by: 按照给定的变量分组,然后按组操作
# mutate: 在当前数据表增加新变量
# 第一步增加每个组的加和,第二步计算比例
data_m <- data_m %>% group_by(variable) %>% mutate(count=sum(value)) %>% mutate(freq=round(100*value/count,2))
再标记相对比例信息
p <- ggplot(data_m, aes(x=variable, y=value, group=gene)) +
geom_bar(stat="identity", position="fill", aes(fill=gene)) +
scale_y_continuous(labels = scales::percent) +
geom_text(aes(label=freq), position=position_fill(vjust=0.5))
p
长矩阵分面绘制
再复杂一些的矩阵 (除了有不同时间点的信息,再增加对照和处理的信息)
library(ggplot2)
library(reshape2)
library(dplyr)
data_ori <- "Gene;Group;Expr;Condition
a;T1;2.6;Control
b;T1;20.8;Control
c;T1;10;Control
d;T1;9;Control
a;T2;2.9;Control
b;T2;9.8;Control
c;T2;11;Control
d;T2;3.3;Control
a;T3;2.1;Control
b;T3;7;Control
c;T3;9.2;Control
d;T3;10.3;Control
a;T4;2;Control
b;T4;3.7;Control
c;T4;12.4;Control
d;T4;11.1;Control
a;T5;2.2;Control
b;T5;19.2;Control
c;T5;9.6;Control
d;T5;10;Control
d;T1;2.6;Treatment
b;T1;20.8;Treatment
c;T1;10;Treatment
a;T1;9;Treatment
d;T2;2.9;Treatment
b;T2;9.8;Treatment
c;T2;11;Treatment
a;T2;3.3;Treatment
a;T3;2.1;Treatment
c;T3;7;Treatment
b;T3;9.2;Treatment
d;T3;10.3;Treatment
a;T4;2;Treatment
c;T4;3.7;Treatment
b;T4;12.4;Treatment
d;T4;11.1;Treatment
a;T5;2.2;Treatment
d;T5;19.2;Treatment
c;T5;9.6;Treatment
b;T5;10;Treatment"
data_m <- read.table(text=data_ori, header=T, sep=";", quote="")
head(data_m)
Gene Group Expr Condition
1 a T1 2.6 Control
2 b T1 20.8 Control
3 c T1 10.0 Control
4 d T1 9.0 Control
5 a T2 2.9 Control
6 b T2 9.8 Control
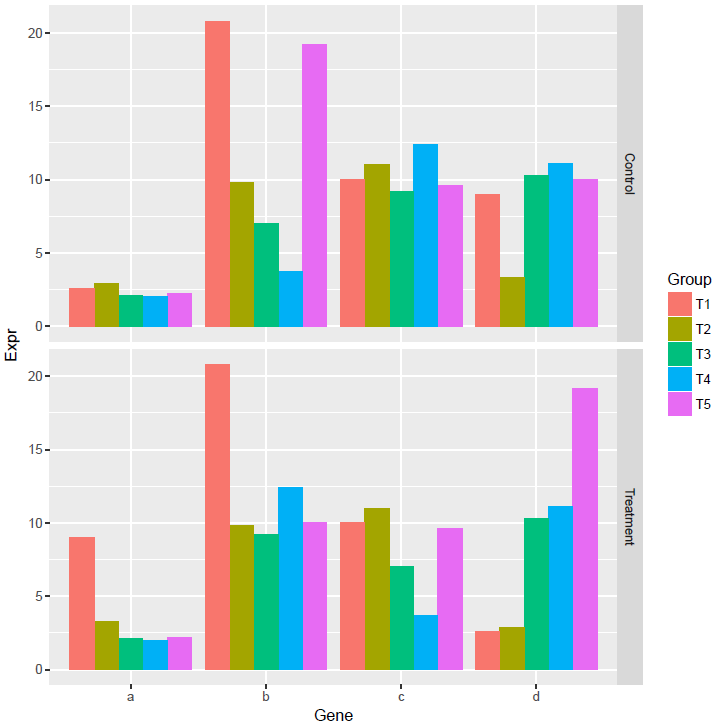
首先看下每个基因在不同组的表达情况, facet_grid和facet_wrap可以对图形分面显示。
# scales: free_y 表示不同子图之间使用独立的Y轴信息
# 但x轴使用同样的信息。
# 其它可选参数有free_x, free, fixed
p <- ggplot(data_m, aes(x=Gene, y=Expr)) +
geom_bar(stat="identity", position="dodge", aes(fill=Group)) +
facet_grid(Condition~., scales="free_y")
p
# 如果没有图形界面,运行下面的语句把图存在工作目录下的Rplots.pdf文件中
#dev.off()
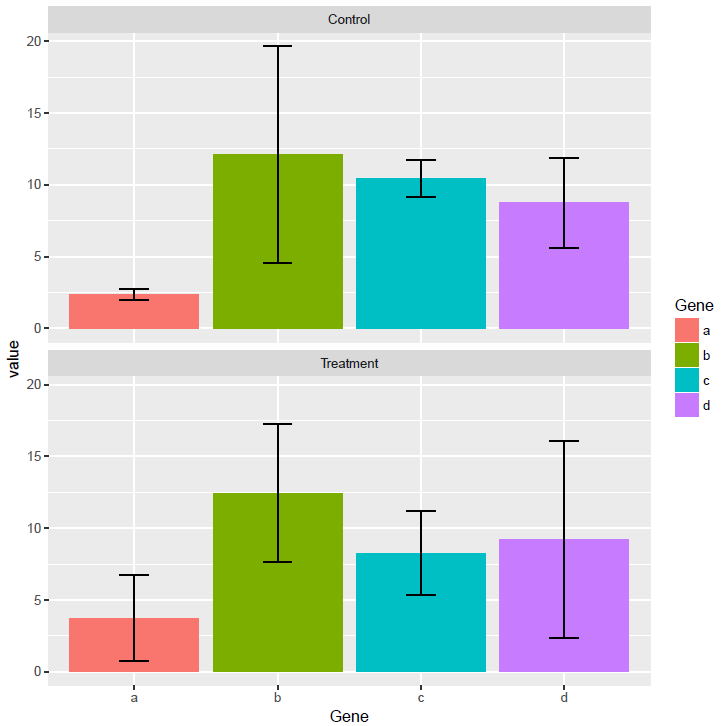
柱子有点多,也可以利用mean±SD的形式展现
# 获取平均值和标准差
# 分组时不只Gene一个变量了,还需要考虑Condition
data_m_sd_mean <- data_m %>% group_by(Gene, Condition) %>% dplyr::summarise(sd=sd(Expr), value=mean(Expr))
data_m_sd_mean <- as.data.frame(data_m_sd_mean)
data_m_sd_mean
Gene Condition sd value
1 a Control 0.3781534 2.36
2 a Treatment 2.9978326 3.72
3 b Control 7.5491721 12.10
4 b Treatment 4.8299068 12.44
5 c Control 1.2837445 10.44
6 c Treatment 2.9458445 8.26
7 d Control 3.1325708 8.74
8 d Treatment 6.8568943 9.22
p <- ggplot(data_m_sd_mean, aes(x=Gene, y=value)) +
geom_bar(stat="identity", aes(fill=Gene)) +
geom_errorbar(aes(ymin=value-sd, ymax=value+sd), width=0.2, position=position_dodge(width=0.75)) +
facet_wrap(~Condition, ncol=1)
p
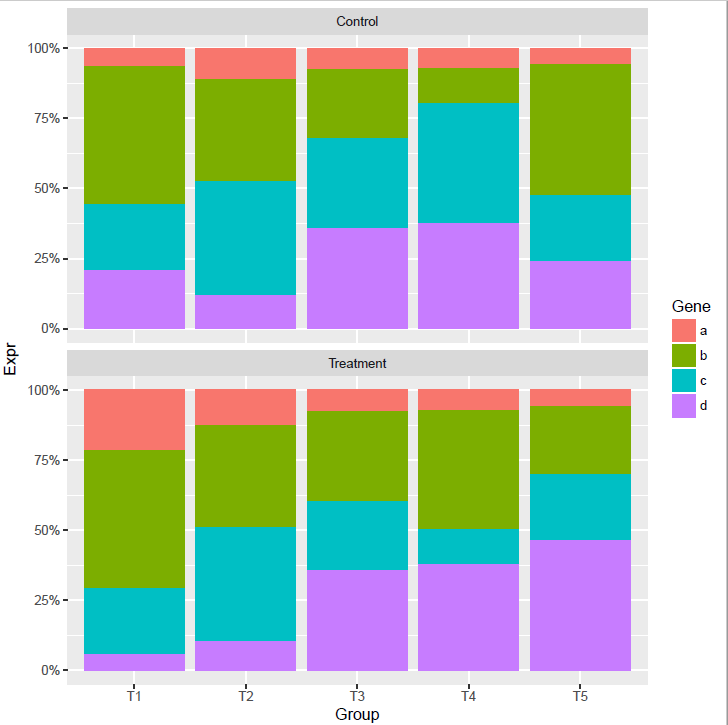
每组里面各个基因的相对表达, 纵轴的显示改为百分比
# position="fill" 展示的是堆积柱状图各部分的相对比例
# position="stack" 展示的是堆积柱状图的原始值,可以自己体现下看卡差别
p <- ggplot(data_m, aes(x=Group, y=Expr)) +
geom_bar(stat="identity", position="fill", aes(fill=Gene)) +
scale_y_continuous(labels = scales::percent) +
facet_wrap(~Condition, ncol=1)
p
facet后,显示正常,不需要做特别的修改
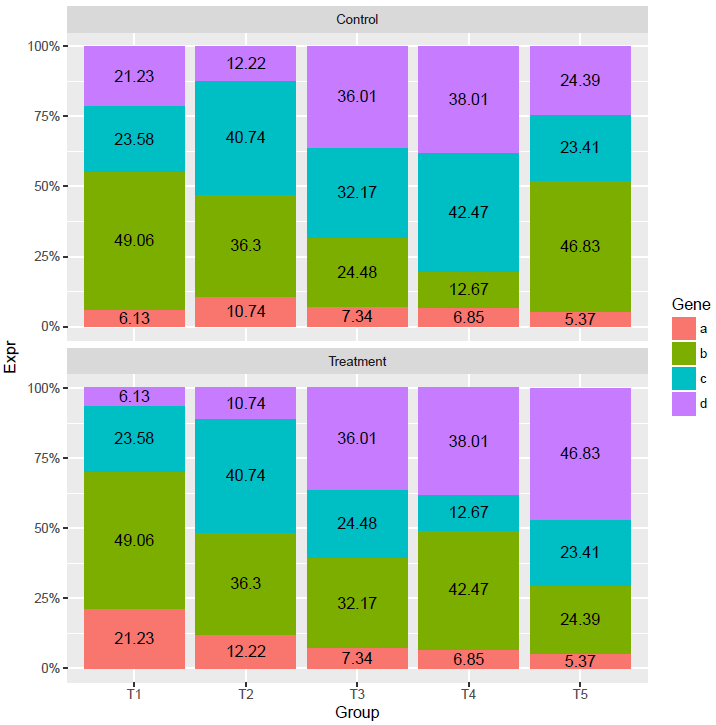
在柱子中标记百分比值 (计算百分比值需要注意了, 文本显示位置还是跟之前一致)
# group_by: 按照给定的变量分组,然后按组操作
# mutate: 在当前数据表增加新变量
# 第一步增加每个组 (Group和Condition共同定义分组)的加和,第二步计算比例
data_m <- data_m %>% group_by(Group, Condition) %>% mutate(count=sum(Expr)) %>% mutate(freq=round(100*Expr/count,2))
p <- ggplot(data_m, aes(x=Group, y=Expr, group=Group)) +
geom_bar(stat="identity", position="fill", aes(fill=Gene)) +
scale_y_continuous(labels = scales::percent) +
geom_text(aes(label=freq), position=position_fill(vjust=0.5)) +
facet_wrap(~Condition, ncol=1)
p
文本显示位置没有问题,但柱子的位置有些奇怪,使得两组之间不可比。
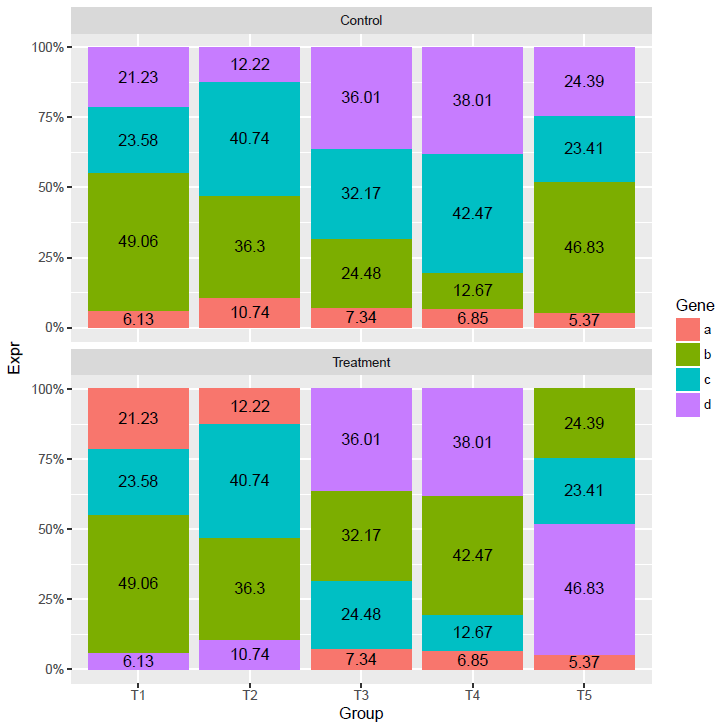
先对数据做下排序,然后再标记文本
# with: 产生一个由data_m组成的局部环境,再这个环境里,列名字可以直接使用
data_m <- data_m[with(data_m, order(Condition, Group, Gene)),]
p <- ggplot(data_m, aes(x=Group, y=Expr, group=Group)) +
geom_bar(stat="identity", position="fill", aes(fill=Gene)) +
scale_y_continuous(labels = scales::percent) +
geom_text(aes(label=freq), position=position_fill(vjust=0.5)) +
facet_wrap(~Condition, ncol=1)
p
这样两种条件下的比较更容易了