前几天,Nature上一篇comment再度引发关于p-value如何使用和解释的文章:Scientists rise up against statistical significance,800多名科学家联合声明拒绝使用基于p-value或置信区间或贝叶斯因子等的二分法将研究结果分为统计显著和统计不显著两个部分,而是应该把置信区间改为兼容性区间, 描述区间所有值的实际含义,尤其是其所代表的的效果 (point estimate)或极值在哪。给定了统计假设,任何极值内的值与研究数据都是兼容的。基于此,作者可以更好的强调数据分析带来的期望值和不确定性,不再对结果过于自信或悲观。
不过一来统计界以后会怎么实施未知,二来签名也未发对p-value的正确使用。那么怎么理解P-value的含义?怎么算是正确使用P-value呢?怎么评估算出的P-value是否正常呢? 就是我们下面要说的。基于传统,后面还是会继续使用显著性这一说法。
统计分析检验获取p-value是我们经常要做的一个工作,比如获得差异基因或富集分析等。通常计算后会得到数百、数千或数万个p-value。考虑到多重假设检验的问题,你可能会想着先做一个校正。
然而,你最先需要做的却是绘制一个直方图。怎么绘制?简单强大的在线绘图-第3版。
在做任何的多重假设检验校正、假阳性率控制或结果解释之前,先绘制这么一个p-value分布直方图,它可以告诉你在所有假设的p值分布,并帮您发现潜在的问题。
p-value分布直方图可能有下面6种可能,我们一一看来。
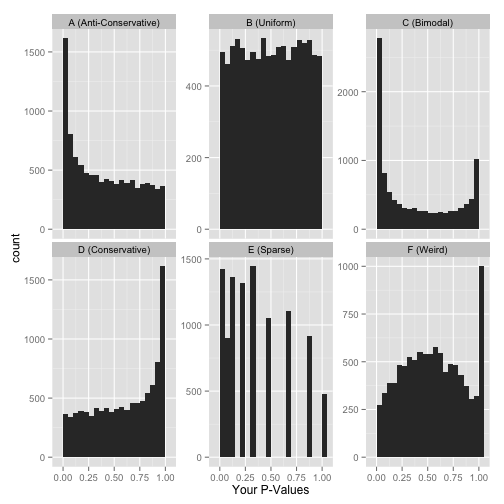
Anti-conservative p-value
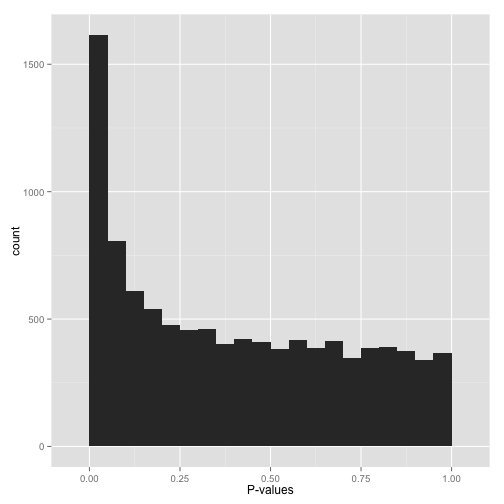
如果p-value分布直方图如上图所示,左侧0值附近有个峰,右侧为近乎均匀分布,那么恭喜你,这是一个很好的分布。
0-1之间均匀分布的p-value代表原假设H0 (null hypothesis)的P值。为什么它们是均匀分布的呢?这是根据p-value的定义来的。在原假设下,p-value有5%的可能低于0.05, 10%的可能低于0.1,以此类推,就是一个均匀分布。
在p-value接近于0值的峰代表的是备择假设H1 (alternative hypothesis) (也包含部分假阳性)。如果把原假设和备择假设分开,p-value的分布应该入下图所示:
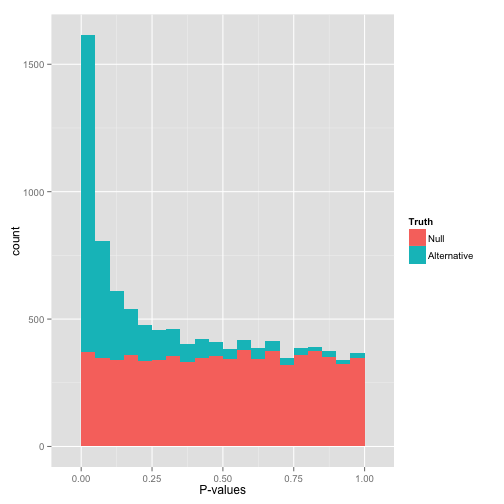
首先可以看到在低p-value处也有一些原假设 (H0),因此不可以简单的说所有p-value<0.05的都是显著的,否则就会获得一些假阳性结果。而且一些备择假设 (H1)的p-value也比较高,这些就是不能通过本次统计检验方法获得的阳性结果,也称为假阴性结果。
多重假设检验校正就是确定显著性的合理阈值。
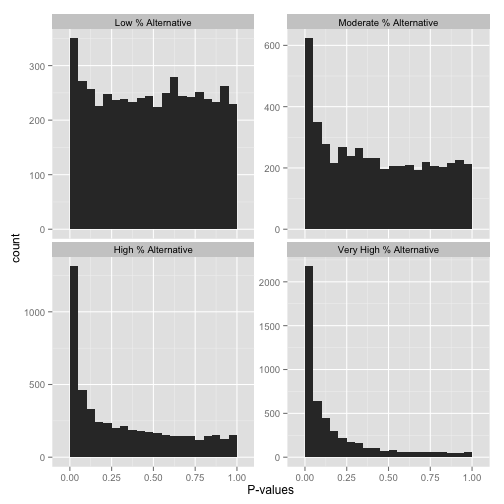
那么怎么判断多少假设是原假设,多少是可以拒绝原假设采用备择假设呢?可以从下面几张图有个直观认识,左侧Peak越高,越多的假设p-value趋近于0, 也就是显著的结果。右侧的柱子越高,更多原假设不能被拒绝。如果想获得定量的评估,可以使用qvalue包。
library(qvalue)
data(hedenfalk)
pvalues <- hedenfalk$p
qobj <- qvalue(p=pvalues)
summary(qobj)
输出不同p-value假设的累计数目
Call:
qvalue(p = pvalues)
pi0: 0.669926
Cumulative number of significant calls:
<1e-04 <0.001 <0.01 <0.025 <0.05 <0.1 <1
p-value 15 76 265 424 605 868 3170
q-value 0 0 1 73 162 319 3170
local FDR 0 0 3 30 85 167 2241
估计原假设 (H0 null hypothesis)的整体比例 (π0),q-value与p-value的关系, qvalue即是定义某一个检验统计显著需要承受的最小假阳性率值。lfdr指在给定的p-value条件下,原假设 (H0)为真的后验概率值。
hist(obj)

均匀分布 Uniform p-value
假如,你的p-value是如下图所示,平平的均匀分布,怎么办呢?
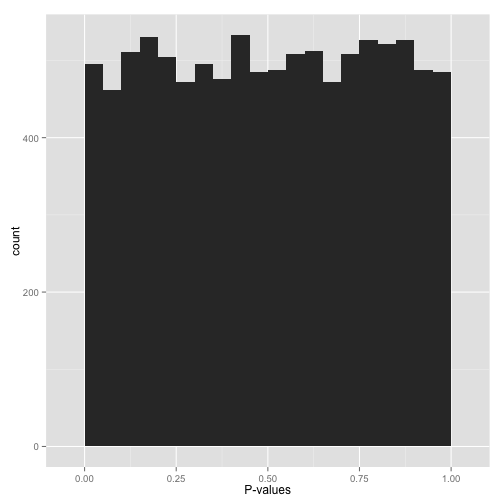
看上去所有的假设都符合原假设,是不是意味着就没有办法拒绝原假设了?其实也不是:
- 起码有一小部分的假设是备择假设,可以用过FDR校正方法如
Benjamini-Hochber等鉴定出来。 - 直接应用
p-value<0.05是不合适的,假阳性率会很高。
双峰 Bimodal p-values
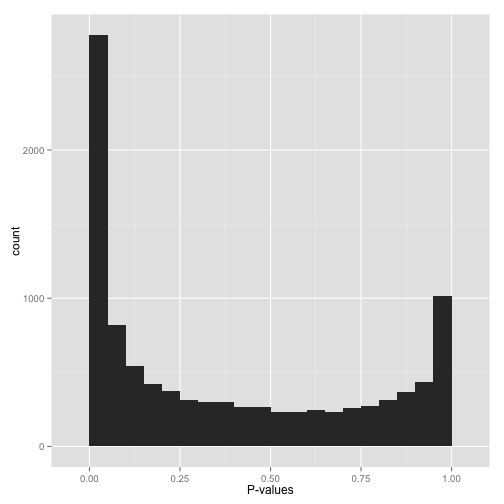
如前面所示在p-value=0处有一个峰,但在p-value=1处也有一个?怎么解释。
首先不要对这些p-value应用假阳性率控制。为什么呢?因为一部分FDR控制算法是基于P-value在1附近是均匀分布的。如果不符合这个前提,计算出的显著性会很少。
下一步找出为什么p-value会有这个分布,针对性解决:
- 是否使用的是单端检验 (
one-tailed test) (如检验药物处理后基因表达上调)。如果是这样,p-value接近1的正好是相反的变化 (如基因表达下调)。如果您同时关注上下调,则采用双端检验 (two-sided test)。如果您不想包含另一种变化,则在检验前先过滤掉这些。(注:比如富集分析时只关注富集) - 是否
pvalue接近1的情况都是病态情况,如基因差异表达分析中,一些软件会赋予在所有样品中都不表达的基因检验pvalue为1,这样的情况直接过滤掉就好。(注:一般分析时是提前过滤。)
Conservative p-values
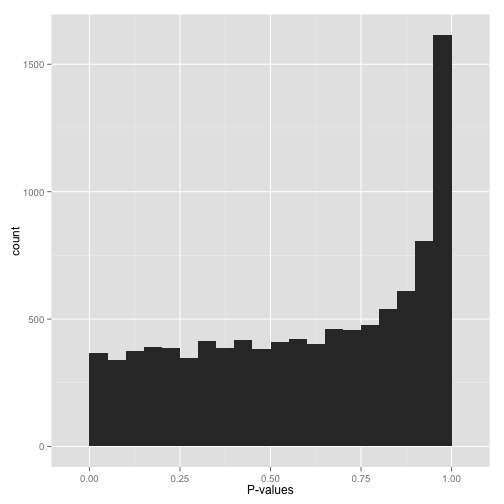
看到这个分布,不要鲁莽的下结论:没有任何统计显著的假设。如果真的没有统计显著性假设,p-value的分布应该是均匀的 Uniform, 这是因为p-value就是这么定义的:原假设下均匀分布。
如果p-value呈现这个分布,说明统计检验使用错了。其原因可能是数据的分布不符合统计检验的假设,比如统计检验适用于连续数据,而提供的是离散数据,或者统计检验适用于正态分布数据,而提供的数据严重不符合等。最好的解决办法是找一个友好的统计学家朋友帮助您。
我们一直强调可视化的是原始p-value的分布,如果使用的工具不小心提供的是校正后的p-value,比如使用Bonferroni correction,那么校正后的p-value可能是这个分布。
稀疏分布 Sparse p-values
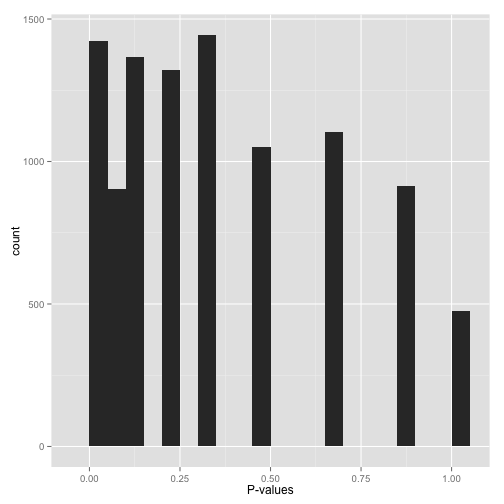
如图所示,获得的p-value的值比较单一,假如做了10,000次统计检验,只获得很少的不同的检验p-value,可以使用下面的代码获取总共有多少不同的p-value。
length(unique(mypvalues))
为什么会获得这样的p-value呢?
- 自展或置换检验 (
bootstrap or permutation test)的迭代次数太少。 - 数据集小的时候运行了非参数检验 (如
Wilcoxon rank-sum test或Spearman correlation),尝试扩大样本量或数据转换为可以进行参数检验。
不要做假阳性率控制,因为p-value的分布不是连续的。
悟空庙宇P-value (“What the…?!?”)
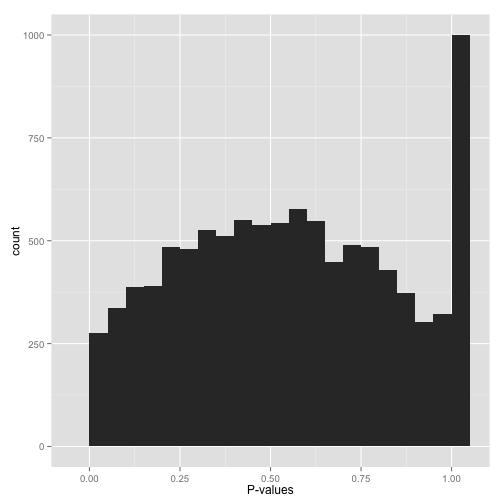
像不像孙悟空变的一座庙,尾巴做旗杆?中间的P-value有个凸起,在1附近有个峰。
最好的方式是求助于统计学家,当然在这之前,看下数据的分布,了解下所用的统计方法,先有个直观认识。
所以p-value不是算出来就可以用了,观察其分布,可以帮助我们判断数据分布是否合适,选用的统计检验方法是否合适,后期如何进行处理,对结果解释增强可信度。
参考
- http://varianceexplained.org/statistics/interpreting-pvalue-histogram/
- http://www.bioconductor.org/packages/release/bioc/vignettes/qvalue/inst/doc/qvalue.pdf
- https://www.nature.com/articles/d41586-019-00857-9
- https://stats.stackexchange.com/questions/10613/why-are-p-values-uniformly-distributed-under-the-null-hypothesis#