Tutorial for smRNA-Seq analysis
Here summarizes the workflow for quantifying the expression of miRNAs using smRNA-Seq data.
File format for smRNA-Seq
Due to the highly redundant of sequenced reads in smRNA-Seq, their data usually saved in a read-tab-count like file as indicated below. Data set used can be downloaded from here.
#SEQUENCE =
#COUNT = counts
SEQUENCE COUNT
TAGCTTATCAGACTGATGTTGACTCGTATGCCGTCT 84080
TAGCTTATCAGACTGATGTTGATCGTATGCCGTCTT 76676
TTAACGCGGCCGCTCTACAATAGTGATCGTATGCCG 75453
AGCGTGTAGGGATCCAAATCGTATGCCGTCTTCTGC 67857
AGCGTGTAGGGATCCAAATCGTATGCCGTCTTCTGT 63472
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA 36186
TGAGGTAGTAGTTTGTGCTGTTTCGTATGCCGTCTT 35574
TGGCTCAGTTCAGCAGGAACAGTTCGTATGCCGTCT 27660Transfer read-tab-count to FASTA file for supplying to quantifier.pl in mirDeep2
Since quantifier.pl requires FASTA file in specific formats, one can use collapsemiRNAreads.py at my Github.
collapsemiRNAreads.py -i GSM416753.txt -H 3 -s 'MEF' >GSM416753.fa
#Currently if you have multiple `read-tab-count` file for multiple samples, you have to transfer them separately with different `three-letter-symbol` given to `-s` to indicate the origin of reads and concatenate each file using `cat` command to one final file. FASTA format:
The name in FASTA file must obey `SSS_INT_xINT` format.
SSS is a three letter code indicating the sample origin
INT is just a running number
xINT is the number of read occurrencesClip adaptor
Once got the reads file, please check if the adaptor is removed. Normally if all reads have same length, we can assume no adaptor-removing is performed. Even unfortunately, researchers normally do not provide adaptor sequences. One can get adaptor sequences by performing multiple sequence aligenment (MSA) of first tens to hundards reads and selectling the end common sequence as adaptor. T-coffee is a great on-line tool to perform MSA.
Once getting the adaptor sequence, one can use fastx_clipper from FASTX tools.
fastx_clipper -a TCGTATGCCGT -l 17 -v -i GSM416732.fa -o GSM416732.clipped.fa
#If there is error like 'invalid quality score value', add '-Q 33' as below
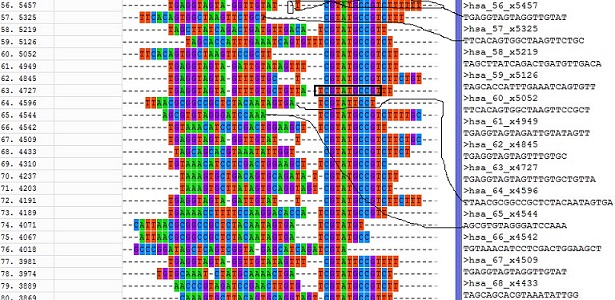
fastx_clipper -a TCGTATGCCGT -l 17 -v -i GSM416732.fa -o GSM416732.clipped.fa -Q 33 Here I used Mega result as an example. The left colorfull letters show the MSA result of 25 reads and black-boxed 11 letters were selected as adaptor sequence. The length of adaptor sequence normally should be larger than 10. The right txt shows the adaptor-clipping result connected by thin-lines.
Quantify miRNA expression
Use the following command to quantify miRNA expression and check your result in file miRNAs_expressed_all_samples_hela.csv.
quantifier.pl -p hsa.hairpin.fa -m hsa.mature.fa -r GSM416732.clipped.fa -t hsa -j -W -y helaTrim reads and quantify miRNA expression again
The 3’ ends of canonical miRNAs are often subject to untemplated additions, especially the 39 ends of mirtron-3p species. Then sometimes we want to trim 3’ end reads one by one and perform mapping process for each trim.
Here I constrcuted a flow to simply the process. All one need is the main program quantifier.sh, and depeneded three programs trimFasta.py, quantifier.modified.pl a modified version of quantifier.pl.
quantifier.sh -p hsa.hairpin.fa -m hsa.mature.fa -r GSM416732.clipped.fa -t hsa -y helaThe principle is like described below. First, map all reads to miRNA precursor using; Second, save mapped reads; Third, extract unmapped reads and trim the 3’ last nucleotide; Forth, map trimmed reads again and save those with no more than 20 mapping loci (this number can be changes as wanted) into mapped reads; Fifth, repeat trimming and mapping process until all reads are shorted than a given length or the cycle-index larger than given number; Sixth, map all saved mapped reads.